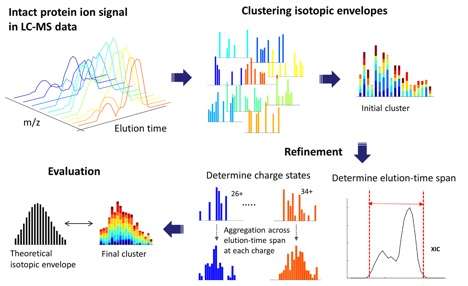
用于分析完整蛋白质的新型开源软件
估计人类基因组中的20,300个基因编码蛋白质。作为完整蛋白质形式的蛋白质本身的数量可能高达10亿。这一大量数字使得人类的功能性蛋白质结构 - 称为蛋白质组 - 比基因组更难以表征。然而,蛋白质组的特征对于理解介导疾病诊断,治疗和预防的蛋白质的活性和功能至关重要。理解存在于人体外的环境中的蛋白质也是必要的。
通常,用于表征蛋白质组的蛋白质组学数据通过液相色谱 -质谱(LC-MS)分析策略收集。这样的仪器旨在通过精确测量电荷,质量和重量来揭示蛋白质的功能和活性。
由主要作者Jungkap Park和太平洋西北国家实验室(PNNL)的科学家们发表的一篇新的“自然方法”论文介绍了Informed-Proteomics,这是一种用于从质谱分析中鉴定完整蛋白质的新型开源软件套件。它包含一整套用于自上而下蛋白质组学的新型软件工具,用于分析完整蛋白质。
通过提供新的LC-MS特征查找算法,新的数据库搜索算法,半自动学习方法和交互式结果查看器,高效且简化的Informed-Proteomics提供了对当前方法的实质性改进。
研究蛋白质的“本土结构”
在传统的“自下而上”蛋白质组学方法中,蛋白质被消化成肽用于质谱鉴定。该方法提供了更高的通量,但是关于完整和活性蛋白质形式的结果可能是不确定的。
自上而下的方法分析每种蛋白质,而分子是完整的。通过这种方式,自上而下的蛋白质组学保留了关于翻译后修饰,同种型和统称为蛋白质形式的分子组合的有价值信息。
共同作者,PNNL综合组学科学家兼团队负责人Sam Payne说:“研究一种蛋白质的天然结构非常重要”,因为有关该蛋白质的更多信息得以保存。然而,他补充说,“整个蛋白质的研究存在非常独特的挑战。”
自上而下的蛋白质组学的技术障碍是“达到你想要的规模,”佩恩说。从自上而下方法得到的光谱要复杂得多,并且需要新的软件工具和新算法来满足他所谓的测量细胞中所有蛋白质的“极具挑战性”的想法。
“自上而下,你所寻求的是非常大的,”佩恩说 - 这需要正确的数学“来组织一种有效的搜索方式。”
“搜索空间”和乳腺癌测试
为什么这么大规模?例如,在自上而下的蛋白质组学中,完整蛋白质的大小意味着电离后的信号在很多维度上展开。另一方面,佩恩所谓的潜在蛋白质的“搜索空间”非常大。蛋白质的组合世界可以达到十亿。
作者通过使用已知具有显着差异的人 - 小鼠异种移植腔和基底乳腺肿瘤样本,与其他几种流行的自上而下蛋白质组学工具一起评估了Informed-Proteomics。
在分析两种乳腺癌亚型中的3,000多种蛋白质形式时,PNNL的作者发现,与最近使用不同方法的自上而下分析相比,他们的新软件工具发现差异表达的蛋白质形式多十倍。
PNNL作者的一个优势来自于PNNL在仪器和信息学方面“在自上而下的领先分析中的悠久历史”,Payne表示,这一事实反映了共同作者Richard D. Smith的工作。“作为一个团队,我们可以在分析的各个方面进行改进,包括计算和技术。”
目前,液相色谱和质谱仪器的数据集质量普遍提高,同时样品处理方案的质量也在不断提高。本文的作者报告说,由于需要处理大量更复杂的自顶向下质谱,因此迫切需要开发用于自信蛋白质鉴定和定量的算法和软件工具。
推荐内容
-
分子谜题揭示了胎儿发育的未知阶段
通过对来自早期小鼠胚胎的单个细胞进行基因分析,瑞典卡罗林斯卡医学院的研究人员发现了以前未知的从受精卵到生命的胎儿发育的细胞阶段...
-
转基因作物的生长在20年内首次放缓
根据周三公布的一项研究,经过20年的稳定增长后,转基因作物的增长率首次下降。在第一个转基因植物上市20年后,2014年全球转基因作物种植面
-
研究发现雄性小鼠体形不大
据西澳大利亚大学研究人员今天发表在英国皇家学会学会B:生物科学的一项新研究,暴露于其他男性竞争对手的雄性小鼠的阴茎骨较厚。博士来自
-
如何修复DNA断裂
来自达姆施塔特工业大学生物系的一组研究人员发现修复DNA损伤的过程远比先前假设的复杂。双螺旋中断的末端不仅仅是连接在一起,它们首先在
-
科学家在“一次性”DNA中发现了未知病毒
机会发现开辟了一种寻找未知病毒的新方法。在发表在病毒进化杂志上的研究中,牛津大学动物学系的科学家们发现,新一代测序及其相关的在...
-
研究提出精神分裂症的第一基因组解释
在一项新的研究中,布法罗大学的遗传学家已经证明了早期发育中重要神经通路的缺陷可能是生命后期精神分裂症发病的原因。科学家们在一个...
-
新研究表明 某些抗生素耐药性感染正在上升
根据急诊医学年鉴(Annals of Emergency Medicine)的最新研究,加利福尼亚州急诊科分析的近6%的尿路感染是在一年的研究期内由耐药细菌引
-
牛奶中新型抗生素抗性基因
伯尔尼大学的研究人员已经从奶牛的细菌中发现了一种新的抗生素抗性基因。该基因赋予对所有β-内酰胺抗生素的抗性,包括用于抗甲氧西...
-
基石药业有望摘下首个国产PD-L1药物
2月26日,专注于开发及商业化创新肿瘤免疫治疗及分子靶向药物的基石药业将正式在香港联交所上市。基石药业IPO的定价为每股12港元,该公司计
-
基因突变涉及罕见的免疫缺陷
一个国际研究小组对来自科威特和沙特阿拉伯两个家庭的儿童的基因组进行了测序,这两个家庭受到基因未确诊的联合免疫缺陷(CID)的影响,这是