科学家提出了一种更快 更准确地研究DNA的算法
来自德国,美国和俄罗斯的科学家团队,包括MIPT生物信息学系主任Mark Borodovsky博士,提出了一种自动搜索基因的算法,使其更有效率。新开发结合了最先进的基因组数据工具的优势。这种新方法将使科学家能够更快,更准确地分析DNA序列,并识别基因组中的全套基因。

尽管描述该算法的论文最近才出现在由牛津期刊出版的生物信息学期刊上,但已经证明该方法非常受欢迎 - 计算机软件程序已被全球1500多个不同的中心和实验室下载。该算法的测试表明它比其他类似算法准确得多。
该发展涉及生物信息学跨学科领域的应用。生物信息学将数学,统计学和计算机科学结合起来研究生物分子,如DNA,RNA和蛋白质结构。DNA基本上是一种信息分子,有时甚至以计算机化的形式描述(见图1),以强调其作为生物记忆分子的作用。生物信息学是一个非常热门的话题; 每个新的测序基因组都会引发许多额外的问题,科学家根本没有时间回答这些问题。因此,自动化流程是任何生物信息学项目成功的关键,这些算法对于解决各种各样的问题至关重要。
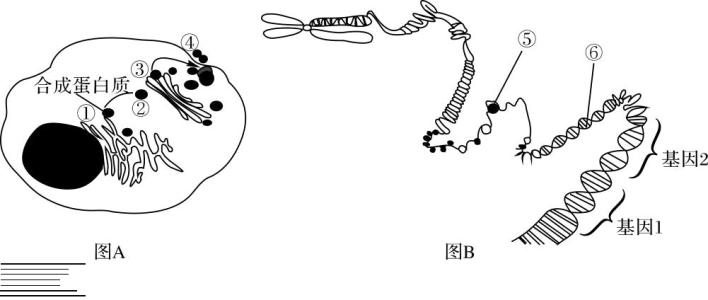
生物信息学最重要的领域之一是注释基因组 - 确定哪些特定的DNA分子用于合成RNA和蛋白质(见图2)。这些部分 - 基因 - 具有重大的科学意义。事实上,在许多研究中,科学家并不需要有关整个基因组的信息(对于单个人类细胞约为2米长),而是关于其信息最丰富的部分 - 基因。通过搜索序列片段和已知基因之间的相似性,或通过检测核苷酸序列的一致模式来鉴定基因部分。该过程使用预测算法来执行。
定位基因切片并非易事,特别是在真核生物中,除了细菌外,其中包括几乎所有广为人知的有机体类型。这是因为在这些细胞中,遗传信息的转移由于编码区(内含子)中的“缺口”而变得复杂,并且因为没有明确的指标来确定区域是否是编码区。
科学家提出的算法确定DNA中哪些区域是基因,哪些区域不是。科学家使用马尔可夫链,这是一系列随机事件,其未来取决于过去的事件。在这种情况下,链的状态是核苷酸或核苷酸词(k-mers)。该算法确定基因组最可能的划分为编码区和非编码区,根据它们编码蛋白质或RNA的能力以最佳方式对基因组片段进行分类。从RNA获得的实验数据给出了额外的有用信息,其可用于训练算法中使用的模型。某些基因预测程序可以使用该数据来提高发现基因的准确性。但是,这些算法需要对模型进行特定类型的训练。对于AUGUSTUS软件程序,例如,具有高水平的准确性,需要训练基因组。这个集合可以使用另一个程序GeneMark-ET获得 - 这是一种自我训练算法。这两种算法结合在BRAKER1算法中,该算法由AUGUSTUS和GeneMark-ET的开发人员联合提出。
BRAKER1已经证明了高效率。已开发的程序已被1500多个不同的中心和实验室下载。该算法的测试表明它比其他类似算法准确得多。BRAKER1在单个处理器上的运行时间为~17.5小时,用于训练和预测长度为120兆碱基的基因组。考虑到使用并行处理器可以显着减少这个时间,这是一个很好的结果,这意味着将来,算法可能更快,通常更有效。
诸如此类的工具解决了各种问题。准确地在基因组中注释基因是非常重要的 - 例如全球1000基因组项目,其初步结果已经发表。该项目于2008年启动,涉及来自75个不同实验室和公司的研究人员。发现了稀有基因变异和基因取代的序列,其中一些可导致疾病。在诊断遗传性疾病时,了解基因切片中的哪些取代会导致疾病发展是非常重要的。该项目绘制了不同人群的基因组图谱,注意到它们的编码区,并鉴定了罕见的核苷酸取代。将来,这将有助于医生诊断心脏病,糖尿病和癌症等复杂疾病。
BRAKER1使科学家能够有效地利用新生物的基因组,加快注释基因组和获取生命科学基本知识的过程。
推荐内容
-
研究人员确定了导致肝纤维化的蛋白质
一个国际科学家团队发现了一种长期寻找的导致肝纤维化(瘢痕形成)的蛋白质,为新疗法铺平了道路。该研究发表在Nature Genetics期刊上。由澳
-
哪些基因对古细菌的能量代谢至关重要
由维也纳大学的Christa Schleper领导的一个研究小组成功地从土壤中分离出了第一个氨氧化古菌:Nitrososphaera viennensis--来自维也纳的
-
科学家们将探索感染流感的老鼠的肺部
在生物化学家和作家艾萨克·阿西莫夫(Isaac Asimov)撰写的1966年小说神奇之旅(Fantastic Voyage)中,为了穿越科学家的身体并将他从脑中的
-
科学家们产生了一种具有半个基因组的新型人类干细胞
来自耶路撒冷希伯来大学,哥伦比亚大学医学中心(CUMC)和纽约干细胞基金会研究所(NYSCF)的科学家们成功地产生了一种新型的胚胎干细胞,它携
-
疟疾寄生虫在骨髓中未被发现累积
一间日疟原虫感染就像一座冰山:这是危险的,部分是因为大部分是隐藏拿出来看。本周在mBio上发表的一项新研究显示,研究人员如何揭示这种寄
-
你需要多少运动才能让你的大脑得到提升
身体活动对身体和心灵都有好处。事实上,医生们早就知道运动会改善思维并减缓认知能力下降的速度,尤其是老年人。但是为了获得更健康的...
-
研究人员发现了调节端粒的机制
染色体的尖端具有称为端粒的结构,与鞋带末端的塑料覆盖物相当。它们起到保护帽的作用,可以防止遗传物质的展开和腐蚀。当端粒不能正常...
-
藻类如何改变其内部太阳能电池板以保持活力
Benning和Kramer实验室之间的合作揭示了大自然在藻类中发现的太阳能电池板如何不断增长和缩小以适应环境的变化,这是确保其宿主保持健康和
-
新的脑相关突变与自闭症谱系障碍有关
一个国际联盟已经确定了与自闭症谱系障碍相关的新突变,提高了我们对该疾病的认识并可能指导治疗方法。该团队包括沙特阿拉伯国王阿卜杜...
-
红海珊瑚是多种细菌的家园
珊瑚礁为大量不同种类的细菌提供了生态位,其中许多细菌被认为可以共生为宿主提供营养。在红海海岸线周围2000公里的珊瑚礁中发现了大约200